
Seaborn Basics (Python)
Published:
Generate a volcano plot using `Python!
Introduction
This document demonstrates how to generate a volcano plot using Python by reading a CSV file that contains gene expression data. The dataset must include at least three mandatory columns:
log2FC(Log2 Fold Change)p_value(P-value for statistical significance)Gene_symbol(or Gene EntrezID or Gene ENSEMBL ID)
Each step is explained in detail, with code chunks for clarity.
Installing Required Libraries
import subprocess
import sys
required_packages = ['pandas', 'matplotlib', 'seaborn']
# Check for missing packages
for pkg in required_packages:
try:
globals()[pkg] = __import__(pkg)
print(f"{pkg} version: {globals()[pkg].__version__}")
except ImportError:
print(f"Error: {pkg} is not installed.")
print(f"Installing {pkg}...")
subprocess.check_call([sys.executable, "-m", "pip", "install", pkg])
globals()[pkg] = __import__(pkg) # Import the package after installation
print(f"{pkg} has been installed.")
print(f"{pkg} version: {globals()[pkg].__version__}")
Import libraries:
import os
import pandas as pd
import seaborn as sns
import matplotlib.pyplot as plt
Setting Up Project Directories
project_dir = "/Users/debojyoti/Projects/seaborn_basics"
data_dir = os.path.join(project_dir, "input_data")
result_dir = os.path.join(project_dir, "results")
os.makedirs(result_dir, exist_ok=True)
Reading the input data using pandas
input_file = os.path.join(data_dir, "test_input_file.csv")
data = pd.read_csv(input_file)
print(data.head())
# Gene_symbol log2FC neg_log10pval log2FC_sq p_value
# 0 Gene7 2.267283 1.725017 5.140572 0.018836
# 1 Gene9 3.027636 3.804565 9.166577 0.000157
# 2 Gene11 1.957304 0.261662 3.831041 0.547442
# 3 Gene12 3.429968 3.787973 11.764681 0.000163
# 4 Gene13 -2.083291 3.899395 4.340102 0.000126
Transforming Data for Visualization
pval_cutoff = 0.05
log2fc_cutoff = 1
data["logP"] = -np.log10(data["p_value"])
data["negLog2FC"] = -data["log2FC"]
conditions = [
(data["p_value"] < pval_cutoff) & (data["negLog2FC"] > log2fc_cutoff),
(data["p_value"] < pval_cutoff) & (data["negLog2FC"] < -log2fc_cutoff),
]
choices = ["Upregulated", "Downregulated"]
data["regulation"] = np.select(conditions, choices, default="Non-significant")
data.head()
# Gene_symbol log2FC neg_log10pval log2FC_sq p_value logP negLog2FC regulation
# 0 Gene7 2.267283 1.725017 5.140572 0.018836 1.725017 -2.267283 Downregulated
# 1 Gene9 3.027636 3.804565 9.166577 0.000157 3.804565 -3.027636 Downregulated
# 2 Gene11 1.957304 0.261662 3.831041 0.547442 0.261662 -1.957304 Non-significant
# 3 Gene12 3.429968 3.787973 11.764681 0.000163 3.787973 -3.429968 Downregulated
# 4 Gene13 -2.083291 3.899395 4.340102 0.000126 3.899395 2.083291 Upregulated
Selecting Top Genes for Labeling
top_n = 5
top_up = data[data["regulation"] == "Upregulated"].nsmallest(top_n, "log2FC")
top_down = data[data["regulation"] == "Downregulated"].nlargest(top_n, "log2FC")
top_genes = pd.concat([top_up, top_down])
# Set the display width to a larger value, e.g., 1000 characters
pd.set_option('display.width', 120)
top_genes.head()
# Gene_symbol log2FC neg_log10pval log2FC_sq p_value logP negLog2FC regulation
# 2440 Gene6948 -6.064915 2.122147 36.783188 0.007548 2.122147 6.064915 Upregulated
# 1724 Gene4951 -4.996103 4.636459 24.961045 0.000023 4.636459 4.996103 Upregulated
# 900 Gene2644 -4.799338 2.081537 23.033642 0.008288 2.081537 4.799338 Upregulated
# 1002 Gene2924 -4.771924 3.505065 22.771257 0.000313 3.505065 4.771924 Upregulated
# 1326 Gene3821 -4.708490 2.552914 22.169875 0.002800 2.552914 4.708490 Upregulated
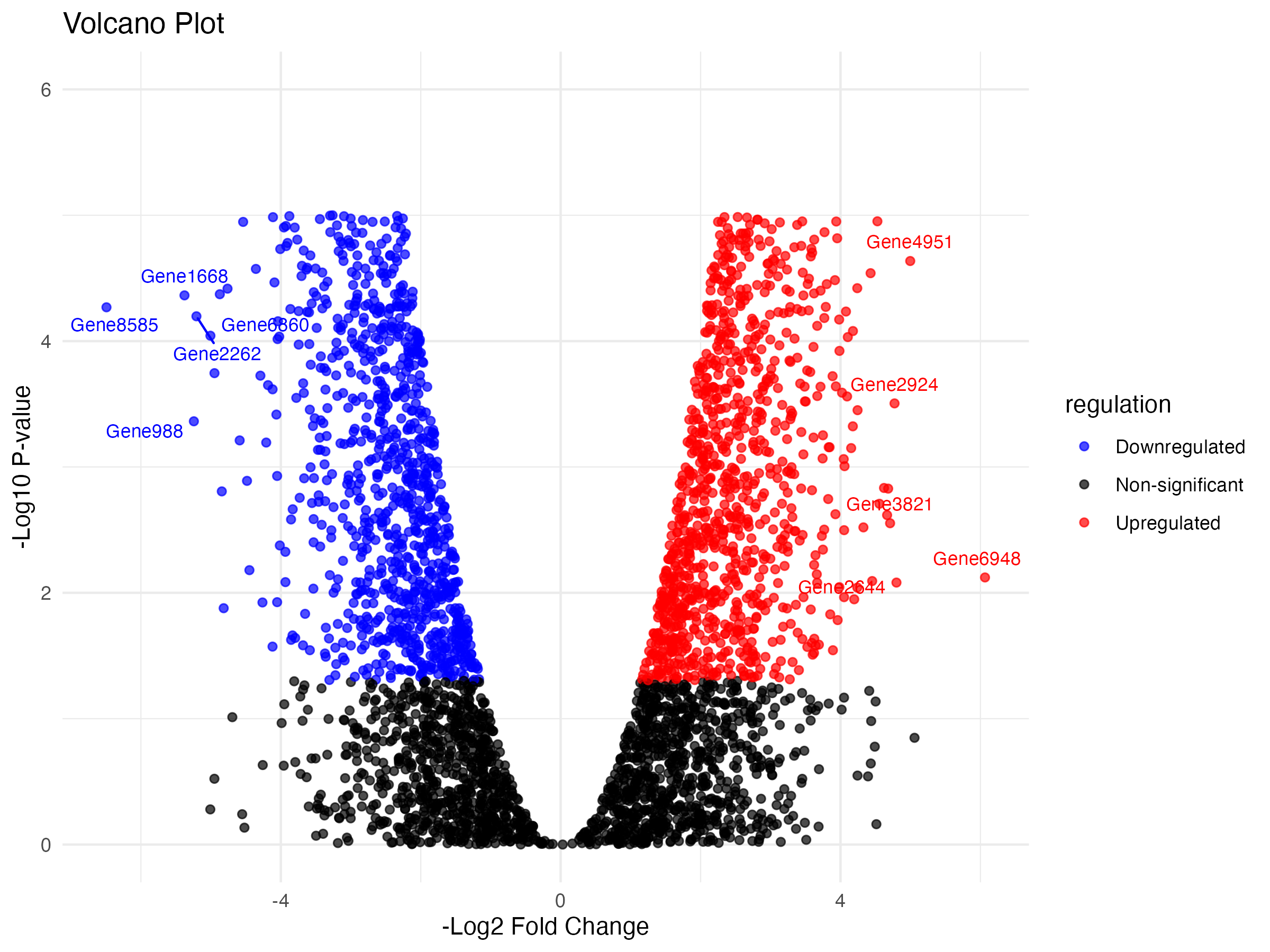
Creating the Volcano Plot
plt.figure(figsize=(8, 6))
sns.scatterplot(
data=data, x="negLog2FC", y="logP", hue="regulation",
palette={"Upregulated": "red", "Downregulated": "blue", "Non-significant": "black"},
alpha=0.7
)
for _, row in top_genes.iterrows():
plt.text(row["negLog2FC"], row["logP"], row["Gene_symbol"], fontsize=8, ha='right')
plt.title("Volcano Plot")
plt.xlabel("-Log2 Fold Change")
plt.ylabel("-Log10 P-value")
plt.ylim(0, 6)
plt.legend(title="regulation")
plt.tight_layout()
plt.show()
Inside the plot (default locations): ‘best’: Automatically chooses the best location (default), ‘upper left’: Legend in the upper-left corner, ‘upper right’: Legend in the upper-right corner, ‘lower left’: Legend in the lower-left corner, ‘lower right’: Legend in the lower-right corner, ‘center left’: Legend in the center of the left side, ‘center right’: Legend in the center of the right side, ‘lower center’: Legend in the center of the bottom side. ‘upper center’: Legend in the center of the top side, ‘center’: Legend in the center of the plot.
Saving the Plot
output_file = os.path.join(result_dir, "volcano_plot.png")
plt.savefig(output_file)
print(f"Plot saved to: {output_file}")
# Plot saved to: /Users/debojyoti/Projects/seaborn_basics/results/volcano_plot.png
<Figure size 640x480 with 0 Axes>
Conclusion
This notebook demonstrated how to load, process, and visualize gene expression data using a volcano plot in Python. We classified genes, highlighted top ones, and saved the plot using seaborn and matplotlib.